|
تضامنًا مع حق الشعب الفلسطيني |
أعران وراثية متعددة
| الأورام العظمية الغضروفية الوراثية المتعددة | |
|---|---|
 صورة لساقين لرجل يبلغ من العمر 26 عامًا يظهر نتوءات متعددة أدت إلى التشوه
| |
| تسميات أخرى | الأعران الوراثية المتعددة |
| تعديل مصدري - تعديل | |
الأورام العظمية الغضروفية الوراثية المتعددة (بالإنجليزية: Hereditary multiple osteochondromas) المعروفة أيضًا باسم الأعران الوراثية المتعددة، هو اضطراب يتميز بتكون كتل أو أورام عظمية متعددة حميدة (أعران) في نهايات العظام الطويلة للأطراف السفلية مثل عظم الفخذ وعظام الظنبوب (قصبة الساق) والأطراف العلوية مثل عظام العضد والساعد. تعرف أيضًا كورم عظمي غضروفي. قد تحدث في أماكن أخرى على العظام المسطحة مثل عظم الحوض والكتف. عدد وتوزيع هذه الأعران يختلف بشكل واسع بين الأفراد المصابين. الأعران عادة ما تكون موجودة خلال مرحلة الطفولة. يصبح الأمر ملاحظًا سريريًا لدى الغالبية العظمى من الأفراد المتضررين عند بلوغهم سن المراهقة.[1][2] وهناك نسبة صغيرة من الأفراد المتضررين معرضين لخطر التحول لورم خبيث ويسمى بالساركومة. يبلغ معدل حدوث حالات الأعران الوراثية المتعددة حوالي واحد من بين كل 50000 شخص.[3] الورم العظمي الغضروفي المتعدد هو المصطلح المفضل الذي تستخدمه منظمة الصحة العالمية.
المظهر
قد يكون أول عرض يظهر عبارة عن ورم يظهر على أحد الأطراف. يمكن أن تحدث تشوهات متعددة، وهي تشوهات حول المستوى الإكليلي الركبتين والكاحلين والكتفين والمرفقين والمعصمين. على سبيل المثال الركبة الروحاء، الكاحل الأروح، انحناء الزند وقصره، ويواجه الكوع المسحوب. غالبية الأفراد المصابين لديهم أورام عظمية غضروفية تظهر حول الركبة. وقد يصاحبة الساعد في HMO.[1][4] علاوة على ذلك قد يحدث قصر القامة ومظهر غير متجانس بشكل عام. عادةً ما تنجم هذه المظاهر عن اضطراب في النمو النمائي خاصة أن الأورام العظمية الغضروفية تنشأ عادةً عند الأطراف الكردوسية (metaphyseal) للعظام الطويلة بالقرب من الأنمية (physis). الورم العظمي الغضروفي المفصلي للورك يمكن أن يؤدي إلى الحد من نطاق الحركة وآلام المفاصل وخلل التنسج الحقي (acetabular dysplasia).[2] وبالمثل يمكن أن يحدث ألم المفاصل في أماكن أخرى ويحدث انضغاط للأوعية الدموية العصبية. علاوة على ذلك يمكن أن يكون العجز الوظيفي فيما يتعلق بأنشطة الحياة اليومية علامة. ألم التشوه الفقري أو التسوية العصبية يجب أن يثير الشكوك حول تورط الفقرات.[3]
علاقة ممكنة بمرض التوحد
لاحظ بعض آباء الأطفال الذين يعانون من HME أنهم يعانون مشاكل اجتماعية تشبه مرض التوحد. لاستكشاف هذه الملاحظات بشكل أعمق قامت دراسة في عام 2012 أجراها معهد سانفورد-بورنهام للأبحاث الطبية باستخدام نموذجًا لفأر مصاب بـ HME لمراقبة الوظيفة الإدراكية. أشارت النتائج إلى أن الفئران المتحورة أظهرت ثلاث خصائص للتوحد: ضعف اجتماعي، ضعف في النطق بالموجات فوق الصوتية، والسلوك المتكرر.[5]
علم الوراثة
HME هو اضطراب وراثي سائد. هذا يعني أن المريض المصاب بـ HME لديه فرصة بنسبة 50% في نقل هذا الاضطراب إلى أبنائه. معظم الأفراد المصابين بـ HME لديهم أحد الوالدين مصاب بهذه الحالة، ولكن حوالي 10% -20% من الأفراد المصابين بـ HME لديهم الحالة نتيجة لطفرة تلقائية، وبالتالي فإن أول شخص في عائلته يتأثر.
تم ربط HME حتى الآن بطفرات في ثلاثة جينات.
- EXT1 الذي يعين كروموسوم 8q24.1[6]
- EXT2 الذي يعين لـ 11p13[7]
- EXT3 الذي يعين الذراع القصير لكروموسوم 19 (رغم أن موقعه الدقيق لم يتحدد بعد بدقة)[8]
تؤدي الطفرات في هذه الجينات عادة إلى تخليق بروتين EXT مقطوع لا يعمل بشكل طبيعي. من المعروف أن بروتينات EXT تعد إنزيمات مهمة في تخليق كبريتات الهيباران (كبريتات الهيبارين). ومع ذلك فإن الآلية الدقيقة التي يتم من خلالها التصنيع المعدل لكبريتات الهيباران التي يمكن أن تؤدي إلى نمو العظام الغير طبيعي المرتبط بـ HME غير واضحة. يُعتقد أن تكاثر الخلية الغضروفية العادي وتمايزها قد يتأثران، مما يؤدي إلى نمو غير طبيعي للعظام.[9][10] نظرًا لأن جينات HME تشارك في تخليق غليكان (كبريتات الهيباران)، يمكن اعتبار HME اضطرابًا خلقيًا في الغلكزة وفقًا لمصطلح CDG الجديد المقترح في عام 2009.[11]
بالنسبة للأفراد الذين يعانون من HME والذين يفكرون في إنشاء أسرة، فإن الاختبارات الجينية والتشخيص قبل الولادة متاح لتحديد ما إذا كان طفلهم الذي لم يولد بعد ورث المرض. يمتلك HME نسبة اختراق تصل 96٪، مما يعني أنه إذا تم نقل الجين المصاب بالفعل إلى الطفل فسيحصل الطفل على ظهور 96٪ للمرض فعليًا، و4% فرصة للإصابة بالمرض ولكن دون إظهاره أبدًا. الرقم 96% يأتي من دراسة واحدة فقط.[12] لاحظت دراسات أخرى كلاً من الاختراق غير الكامل والمتغير ولكن دون حساب النسبة المئوية.[13] في كلتا الدراستين المذكورتين أعلاه كان الأفراد الذين لا يحملون أعراضًا والذين يحملون الجين المعيب من الإناث في الغالب، مما أدى إلى تكهنات بأن الاختراق غير المكتمل من المرجح أن يظهر في الإناث. في الواقع أظهرت دراسات أخرى أن الأولاد/الرجال يميلون إلى الإصابة بالمرض أسوأ من الإناث، وكذلك أن عدد الأعران لدى الأفراد المصابين من نفس العائلة يمكن أن يختلف اختلافًا كبيرًا.[14] من الممكن أيضًا أن تتأثر الإناث بشدة. تختلف شدة الأعراض بين الأفراد حتى في نفس العائلة.
من المرجح أن تكون الأعراض شديدة إذا كانت الطفرة موجودة في جين ext1 بدلاً من ext2 أو ext3؛ ext1 هو أيضًا الجين الأكثر تضررًا في مرضى هذا الخلل.[14]
الفيزيولوجيا المرضية
يتميز بنمو أورام العظم الحميدة المغطاة بالغضاريف حول مناطق النمو العظمي النشط، لا سيما كردوس العظام الطويلة. عادة ما يتم العثور على خمسة أو ستة أعران في الأطراف العلوية والسفلية. المواقع الأكثر شيوعًا هي:[15]
- أقصى عظم الفخذ (70%)
- أدنى الظنبوب (70%)
- عظم العضد (50%)
- أدنى عظم الشظية (30%)
HME يمكن أن يؤدي إلى تقصير وانحناء العظام. الأفراد المصابون غالبًا ما يكون لديهم قامة قصيرة. اعتمادًا على موقعها يمكن أن تتسبب الأعران الخارجية في حدوث المشاكل التالية: ألم أو تنميل من انضغاط الأعصاب، عدم تساوي طول الأطراف، تهيج الأوتار والعضلات، تشوه مفصل الرسغ[16] وكذلك مجموعة محدودة من الحركة في المفاصل المصابة. يعاني الشخص المصاب بـ HME من زيادة خطر الإصابة بنوع نادر من سرطان العظام يُسمى chondrosarcoma عند البلوغ. قد تكون هناك مشاكل في الحياة لاحقًا وقد تشمل ضعف العظام وتلف الأعصاب.[17][18][19] يتراوح معدل التحول المبلغ عنه من 0.57% إلى 8.3% من المصابين بـ HME.[20]
التشخيص
يعتمد تشخيص HMO على إنشاء ارتباط دقيق بين السمات السريرية المذكورة أعلاه وسمات الصور الإشعاعية المميزة. يمكن أن يوفر تاريخ العائلة دليلًا مهمًا للتشخيص. يتم استكمال هذا عن طريق اختبار الجينين حيث من المعروف أن المتغيرات المسببة لHMO هما EXT1 وEXT2. مزيج من تحليل التسلسل وتحليل الحذف لمناطق الترميز بأكملها لكل من EXT1 وEXT2 يكشف المتغيرات المسببة للأمراض في 70% -95% من الأفراد المتضررين.[3][4] السمة المميزة للتشخيص الشعاعي هي وجود أورام عظمية غضروفية في النهايات كردوس للعظام الطويلة التي تمثل فيها القشرة النخاعية والنخاع العظمي امتدادًا مستمرًا للعظم المضيف. يتضح هذا بسهولة في الصور الشعاعية للركبتين.[1]
العلاج
مؤشرات التدخل الجراحي في الأفراد الذين يعانون من HMO لا تزال غير واضحة وتختلف بشكل كبير عبر المواد الطبية. يتضمن العلاج الجراحي العام لـ HMO إجراءً واحدًا أو أكثر من الإجراءات التالية: استئصال الورم العظمي الغضروفي، إطالة العظم تدريجيًا أو بشكل حادًا مثل إطالة الزند، قطع العظم، تَثْبيتُ المُشاشَة الجزئي المؤقت.[1][3] ومع ذلك هناك القليل من الأدلة تدعم الممارسة المستمرة لجراحة العظام لدى الأطفال في الورم العظمي الغضروفي المتعدد الوراثي. وجدت المراجعات المنهجية الحديثة أدلة غير كافية لإثبات أن العلاج الجراحي المستمر لـ HMO يحسن الوظيفة إلى حد كبير أو لإثبات أنه يؤثر على نوعية حياة الأطفال المصابين.[2] لتعزيز كمية الأدلة في المواد الطبية وضعت بعض التوصيات. يوجد طلب كبير لبناء دراسات مستقبلية يمكن أن توفر علاقة أكثر وضوحا بين العمليات الجراحية وخصائص المريض ونتائجها.حيث أنه بتصاميم الدراسة الحالية سوف تستمر في طرح المزيد من الأسئلة بدلا عن الإجابات. تم استخدام رأب مفصل الورك الكلي لعلاج HMO الحاد والألم في مفصل الورك. يمثل رأب مفصل الورك الكلي للأفراد المصابين بـ HMO تحديًا بسبب تشوه التشريح والعمليات الجراحية المتكررة التي يتم إجراؤها لمعالجة الشكاوى المتعلقة بالتخثر.[21]
علم الأوبئة
يقدر أن حدوث HME في 1 من كل 50000 شخص.[15][19]
صور إضافية
-
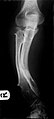
الورم العظمي الغضروفي المتعدد يسبب تشوه في الساعد.
-

ورم عظمي غضروفي متعدد في الحوض
-

ورم عظمي غضروفي متعدد حول الركبة
-

أشعة مقطعية لورم عظمي غضروفي
.jpg)
.jpg)


المراجع
- ^ أ ب ت ث EL-Sobky، TA؛ Samir، S؛ Atiyya، AN؛ Mahmoud، S؛ Aly، AS؛ Soliman، R (21 مارس 2018). "Current paediatric orthopaedic practice in hereditary multiple osteochondromas of the forearm: a systematic review". Sicot-J. ج. 4: 10. DOI:10.1051/sicotj/2018002. PMID:29565244.
{{استشهاد بدورية محكمة}}: الوسيط غير المعروف|PMCID=تم تجاهله يقترح استخدام|pmc=(مساعدة) - ^ أ ب ت Makhdom، AM؛ Jiang، F؛ Hamdy، RC؛ Benaroch، TE؛ Lavigne، M؛ Saran، N (20 مايو 2014). "Hip joint osteochondroma: systematic review of the literature and report of three further cases". Advances in Orthopedics. ج. 2014: 180254. DOI:10.1155/2014/180254. PMID:24963411.
{{استشهاد بدورية محكمة}}: الوسيط غير المعروف|PMCID=تم تجاهله يقترح استخدام|pmc=(مساعدة) - ^ أ ب ت ث Wuyts، W؛ Schmale، GA؛ Chansky، HA؛ وآخرون (21 نوفمبر 2013). "Hereditary Multiple Osteochondromas". GeneReviews. University of Washington, Seattle. مؤرشف من الأصل في 2017-01-28. اطلع عليه بتاريخ 2018-03-24.
- ^ أ ب Alvarez، CM؛ De Vera، MA؛ Heslip، TR؛ Casey، B (سبتمبر 2007). "Evaluation of the anatomic burden of patients with hereditary multiple exostoses". Clin Orthop Relat Res. ج. 462: 73–79. DOI:10.1097/BLO.0b013e3181334b51. PMID:17589361.
- ^ "Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate". Proceedings of the National Academy of Sciences of the United States of America. ج. 109 ع. 13: 5052–6. مارس 2012. Bibcode:2012PNAS..109.5052I. DOI:10.1073/pnas.1117881109. PMID:22411800.
{{استشهاد بدورية محكمة}}: الوسيط غير المعروف|PMCID=تم تجاهله يقترح استخدام|pmc=(مساعدة) - ^ "Genetic heterogeneity in families with hereditary multiple exostoses". American Journal of Human Genetics. ج. 53 ع. 1: 71–9. 1993. PMID:8317501.
{{استشهاد بدورية محكمة}}: الوسيط غير المعروف|PMCID=تم تجاهله يقترح استخدام|pmc=(مساعدة) - ^ "Assignment of a second locus for multiple exostoses to the pericentromeric region of chromosome 11". Human Molecular Genetics. ج. 3 ع. 1: 167–71. 1994. DOI:10.1093/hmg/3.1.167. PMID:8162019.
- ^ "A gene for hereditary multiple exostoses maps to chromosome 19p". Human Molecular Genetics. ج. 3 ع. 5: 717–22. 1994. DOI:10.1093/hmg/3.5.717. PMID:8081357.
- ^ "Hereditary multiple exostoses and heparan sulfate polymerization". Biochimica et Biophysica Acta. ج. 1573 ع. 3: 346–55. 2002. DOI:10.1016/S0304-4165(02)00402-6. PMID:12417417.
- ^ "Manifestations of hereditary multiple exostoses". The Journal of the American Academy of Orthopaedic Surgeons. ج. 13 ع. 2: 110–20. 2005. DOI:10.5435/00124635-200503000-00004. PMID:15850368.
- ^ "CDG nomenclature: time for a change!". Biochim. Biophys. Acta. ج. 1792 ع. 9: 825–6. 2009. DOI:10.1016/j.bbadis.2009.08.005. PMID:19765534.
{{استشهاد بدورية محكمة}}: الوسيط غير المعروف|PMCID=تم تجاهله يقترح استخدام|pmc=(مساعدة) - ^ "Incomplete penetrance and expressivity skewing in hereditary multiple exostoses". Clinical Genetics. ج. 52 ع. 1: 12–6. يوليو 1997. DOI:10.1111/j.1399-0004.1997.tb02508.x. PMID:9272707.
- ^ "Novel mutations in the EXT1 gene in two consanguineous families affected with multiple hereditary exostoses (familial osteochondromatosis)". Clinical Genetics. ج. 66 ع. 2: 144–51. أغسطس 2004. DOI:10.1111/j.1399-0004.2004.00275.x. PMID:15253765.
- ^ أ ب "Severity of disease and risk of malignant change in hereditary multiple exostoses. A genotype-phenotype study". The Journal of Bone and Joint Surgery. British Volume. ج. 86 ع. 7: 1041–6. سبتمبر 2004. DOI:10.1302/0301-620x.86b7.14815. PMID:15446535. مؤرشف من الأصل في 2020-05-05.
- ^ أ ب Turek's orthopaedics principles and their application (ط. 6th). Philadelphia: Lippincott Williams & Wilkins. 2005. ص. 263. ISBN:9780781742986.
- ^ Davies، A. Mark؛ Pettersson، Holger (2002). Pettersson، Holger؛ Ostensen، Harald (المحررون). Radiography of the Musculoskeletal System (PDF). Geneva: World Health Organization. ص. 177, 189. ISBN:978-92-4-154555-6. مؤرشف من الأصل (PDF) في 2014-11-27.
- ^ CANNON JF (1954). "Hereditary multiple exostoses". American Journal of Human Genetics. ج. 6 ع. 4: 419–25. PMID:14349947.
{{استشهاد بدورية محكمة}}: الوسيط غير المعروف|PMCID=تم تجاهله يقترح استخدام|pmc=(مساعدة) - ^ McBride WZ (سبتمبر 1988). "Hereditary multiple exostoses". American Family Physician. ج. 38 ع. 3: 191–2. PMID:3046271.
- ^ أ ب "The natural history of hereditary multiple exostoses". The Journal of Bone and Joint Surgery. American Volume. ج. 76 ع. 7: 986–92. يوليو 1994. DOI:10.2106/00004623-199407000-00005. PMID:8027127. مؤرشف من الأصل في 2020-05-05.
- ^ "Chondrosarcoma in a family with multiple hereditary exostoses". The Journal of Bone and Joint Surgery. British Volume. ج. 82 ع. 2: 261–6. مارس 2000. DOI:10.1302/0301-620X.82B2.0820261. PMID:10755438. مؤرشف من الأصل في 2020-05-05.
- ^ Vaishya، R؛ Swami، S؛ Vijay، V؛ Vaish، A (5 يناير 2015). "Bilateral total hip arthroplasty in a young man with hereditary multiple exostoses". BMJ Case Rep. ج. 2015: bcr2014207853. DOI:10.1136/bcr-2014-207853. PMID:25564594.
{{استشهاد بدورية محكمة}}: الوسيط غير المعروف|PMCID=تم تجاهله يقترح استخدام|pmc=(مساعدة)
| أعران وراثية متعددة في المشاريع الشقيقة: | |